- Research article
- Open access
- Published:
A sequence-based approach to identify reference genes for gene expression analysis
BMC Medical Genomics volume 3, Article number: 32 (2010)
Abstract
Background
An important consideration when analyzing both microarray and quantitative PCR expression data is the selection of appropriate genes as endogenous controls or reference genes. This step is especially critical when identifying genes differentially expressed between datasets. Moreover, reference genes suitable in one context (e.g. lung cancer) may not be suitable in another (e.g. breast cancer). Currently, the main approach to identify reference genes involves the mining of expression microarray data for highly expressed and relatively constant transcripts across a sample set. A caveat here is the requirement for transcript normalization prior to analysis, and measurements obtained are relative, not absolute. Alternatively, as sequencing-based technologies provide digital quantitative output, absolute quantification ensues, and reference gene identification becomes more accurate.
Methods
Serial analysis of gene expression (SAGE) profiles of non-malignant and malignant lung samples were compared using a permutation test to identify the most stably expressed genes across all samples. Subsequently, the specificity of the reference genes was evaluated across multiple tissue types, their constancy of expression was assessed using quantitative RT-PCR (qPCR), and their impact on differential expression analysis of microarray data was evaluated.
Results
We show that (i) conventional references genes such as ACTB and GAPDH are highly variable between cancerous and non-cancerous samples, (ii) reference genes identified for lung cancer do not perform well for other cancer types (breast and brain), (iii) reference genes identified through SAGE show low variability using qPCR in a different cohort of samples, and (iv) normalization of a lung cancer gene expression microarray dataset with or without our reference genes, yields different results for differential gene expression and subsequent analyses. Specifically, key established pathways in lung cancer exhibit higher statistical significance using a dataset normalized with our reference genes relative to normalization without using our reference genes.
Conclusions
Our analyses found NDUFA1, RPL19, RAB5C, and RPS18 to occupy the top ranking positions among 15 suitable reference genes optimal for normalization of lung tissue expression data. Significantly, the approach used in this study can be applied to data generated using new generation sequencing platforms for the identification of reference genes optimal within diverse contexts.
Background
Gene expression profiling, including quantitative RT-PCR (qPCR) and microarray experimentation, is invaluable for the molecular analysis of biological systems. The interpretation of results from such experiments (i.e., the determination of differential expression for a particular gene among datasets) is strongly influenced by the selection of reference genes for normalization across datasets [1]. Specifically, gene expression is normalized within a given dataset by calculating the transcript abundance of the gene of interest relative to a gene that is constantly expressed across independent datasets (termed a "housekeeping" or a "reference" gene), and differential expression between two datasets or samples is determined by calculating the ratio of the normalized expression levels for the gene of interest between the two datasets. Typically, housekeeping genes satisfy the following criteria: they are highly expressed in the cell, the variability in expression between samples is minimal, and the genes' expression is not influenced by the experimental conditions tested [2]. Hence, problems arise when housekeeping genes are selected that do not meet these criteria, as fluctuations in these genes may erroneously influence the data interpretation.
Historically, beta actin (ACTB), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and 18 S rRNA have been routinely used as reference genes for qPCR and microarray data normalization. However, a number of studies have shown that expression of these genes varies considerably depending on the specific tissue type and disease state of the tissue [3–16]. Attempts to achieve more reliable normalization include the spiking of synthetic poly-A RNAs for the analysis of cDNA arrays and northern blots, and the combined use of an oligo-(dT)n primer with an 18 S specific primer for qPCR analysis [17, 18]. In addition, re-mining of large microarray datasets for the identification of novel, highly stable genes, as well as use of a combination of reference genes instead of a single gene for normalization, are some of the other approaches taken to address this problem [11, 13, 19].
Recently, efforts have been made to identify more suitable reference genes for microarray and qPCR studies of lung cancer. Specifically, candidate reference genes have been identified from the mining of microarray gene expression data to identify the least variable genes, followed by validation of expression using qPCR [11, 20, 21]. However, as microarray data do not provide absolute abundance values for transcripts, selection of reference genes from this type of data is inherently problematic. To circumvent this handicap in the utilization of microarray data, we turn to the use of large-scale expression profiling permitted by serial analysis of gene expression (SAGE) experimentation for the identification of novel reference genes optimal for the study of lung cancer. This approach, which we have termed normalization of expression by permutation of SAGE (NEPS), takes advantage of the fact that SAGE is a transcriptome profiling technique that identifies the absolute abundance levels of transcripts by direct enumeration of sequence tag counts, thus allowing the direct comparison of expression levels across multiple profiles without the need for reference or housekeeping genes [22].
NEPS adopts a permutation test approach designed for analyzing relatively small sample sizes, such as those typically encountered with SAGE. Unlike the conventional T-test, the permutation test is non-parametric [23]. The null hypothesis states that the mean gene expression levels in two groups of SAGE libraries being compared (in this case normal and cancer), are the same. For this analysis, samples from both the normal and the cancer groups are pooled, followed by random sampling to create a simulated Group 1 and a simulated Group 2. For each gene, the difference in expression between these two simulated groups was measured. This exercise was repeated 10,000 times, thus generating a simulated mean μ and a simulated standard deviation σ. The permutation score (PS) of a given gene is defined by 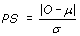 , where O is the true difference between the average expression levels in the two groups. Hence, for a given gene, the closer the permutation score is to zero, the more it satisfies the constancy requirement.
, where O is the true difference between the average expression levels in the two groups. Hence, for a given gene, the closer the permutation score is to zero, the more it satisfies the constancy requirement.
To demonstrate the utility of NEPS for selecting genes that satisfy the constancy requirement, we analyzed 24 bronchial epithelial lung SAGE libraries, 2 lung parenchyma libraries, and 11 lung squamous cell carcinoma libraries. From this analysis, NEPS selected 15 genes, which we hereafter refer to as the lung-NEPS reference genes (Table 1). We further demonstrate that (1) while these genes perform well as reference genes for lung, they are not satisfactory for normalization of expression data from other tissues, suggesting that reference genes are tissue-specific, and (2) in lung cancer datasets, differential gene expression determination and subsequent pathway analyses are improved after normalization using the lung-NEPS reference genes.
Methods
SAGE library construction
26 normal and 11 lung cancer SAGE libraries were constructed and used in the analysis [24]. The construction of the 26 normal libraries has been previously described [25, 26]. 24 of these libraries were generated from exfoliated bronchial cells obtained from bronchial brushes, and two libraries from normal lung parenchyma (Additional file 1). Conversely, the 11 cancer libraries were generated from biopsied specimens with six libraries representing lung squamous cell carcinoma and five libraries representing carcinoma in situ. This data can be found at the GEO database with the following series accession numbers: GSE3707, GSE5473, and GSE7898. All samples were acquired under approval by the University of British Columbia - British Columbia Cancer Agency Research Ethics Board (UBC-BCCA-REB) and all subjects provided written consent.
SAGE data from public domain
Publicly available SAGE data were also used in this analysis, representing both brain and breast cancer. Specifically, six normal and 12 breast cancer libraries (Additional file 2) and 7 normal and 19 brain cancer libraries were used (Additional file 3). The libraries were obtained from the cancer genome anatomy project (CGAP) database http://cgap.nci.nih.gov[27, 28].
Permutation test
Given that SAGE libraries are expensive to generate, the number of libraries in a given study is typically small (i.e., in 10's, rather than in 100's). The permutation test is a non-parametric test, which does not assume any underlying distribution. The number of samples required for the test to achieve sufficient statistical power is relatively low compared to other statistical tests (e.g., t-test and χ2-square test). Furthermore, each additional sample increases the power of the test exponentially. The permutation test is a test of the means between two different distributions. Without loss of generality, let us assume that one distribution is for the gene expression level of a particular gene in normal tissues (i.e. subscript n), and that the other distribution is for cancerous tissues (i.e. subscript c). Genes are selected using the following hypotheses:

If there is little difference between the two means, it would make no difference if we mix the cancerous samples with the normal samples. But, if the null hypothesis is rejected, it indicates that the gene expression levels of normal and cancer samples are sufficiently different (the alternative hypothesis). In the following, we show our specific implementation of the test. Let n and c be the number of normal tissue samples and the number of cancerous tissue samples respectively.
-
A.
For each gene, select all the gene-specific normalized tag counts from the normal libraries and all the gene-specific normalized tag counts from the cancer libraries.
-
B.
Randomly select n counts to create a simulated normal set, and calculate the simulated normal mean μ sn .
-
C.
Similarly, select the remaining c counts form the simulated cancerous set. Calculate the simulated cancer mean μ sc .
-
D.
Consider the random variable v = μ sc - μ sn , called the simulated difference.
-
E.
Repeat the steps A to D above m times. Let μ and σ denote the mean and the standard deviation of v.
-
F.
Now separate the libraries back into their true identity: normal or cancerous. Calculate the true observed difference O = μ rc - μ rn , where μ rc denotes the true mean count of the cancerous libraries, and μ rn denotes the true mean of the normal libraries.
-
G.
Calculate the Permutation Score PS where
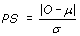 .
. -
H.
Repeat all the above steps for each gene. Sort the permutation score in descending order.
The permutation score is one way to measure how likely the actual observed difference occurs by chance. It is based on standardization, i.e., subtracting the mean and then divided by the standard deviation. The more the true observed difference is from the average (expressed as multiples of the standard deviation), the less likely that the true observed difference is a coincidence. That is to say, the larger the permutation score, the more significant is the observed difference between cancerous and normal samples.
On the other hand, for the sake of evaluating the constancy requirement, the ideal reference gene would have a permutation score equal to 0. This means that there is no difference in the distributions of expression levels between cancerous and normal samples. For the results reported here, we used m = 10,000 permutations.
Data pre-processing
Raw tag counts for each SAGE library were normalized to tags per million (TPM) to facilitate adequate comparison among libraries. Tag-to-gene mapping was performed using the February 5th, 2007 version of SAGEGenie [27]. In cases where multiple SAGE tags mapped to the same gene, the tags were collapsed to capture all potential transcript variants, and a cumulative tag count was utilized for analysis.
Statistical criteria for reference gene selection
The permutation test outlined above was used to identify genes which were statistically similar when comparing the libraries from normal tissue (bronchial epithelium and lung parenchyma) and cancerous tissue of the lung. Three main criteria were used for reference gene selection: permutation score (described above) ≤ 0.15; at least two SAGE tags observed in each library; and an overall average count of ≥ 25 across all samples. For the analysis in brain and breast tissue, the first two criteria were maintained, but due to the lower sequencing depth, an average count of ≥ 10 across all samples was used instead.
Quantitative RT-PCR validation in clinical lung cancer specimens
One microgram of total RNA from 15 lung tumor and matched non-malignant parenchyma samples were converted to cDNA using the High-Capacity cDNA archive kit (Applied Biosystems Inc., Foster City CA). One hundred nanograms of cDNA were utilized for qPCR using the TaqMan Gene Expression Assay (Applied Biosystems Inc). All fifteen lung NEPS genes and six additional reference genes were assayed. All TaqMan probes were pre-optimized by Applied Biosystems. Primer IDs for all genes are provided in Additional file 4. The 30 samples were assayed in triplicate in parallel along with negative (no cDNA template) controls using the 7500 Fast Real-Time PCR System. Appropriate cDNA dilutions were used such that the exponential phase of the amplification curves were within the 40 PCR cycles recommended by the manufacturer (i.e. ranging from 16-36 cycles for the 20 genes and 1-13 cycles for 18SRNA). Cycle thresholds were determined from amplification curves using 7500 Fast System software.
For the analysis of qPCR data, three different methods were used. Within each method, all genes were ranked from best to worst. Subsequently, for each gene, a cumulative ranking across all three methods was determined by summing its rank from each individual method. Two previously published methods, geNorm [14] and NormFinder [29], and the variance of cycle threshold difference (dCt) across all 15 tumor/matched non-malignant sample pairs were the approaches used to determine constancy.
Analysis of publicly available microarray datasets
Lung NEPS genes were used to re-normalize two publicly available microarray datasets. Microarray data were obtained from GEO at NCBI under accession numbers GSE10072 [30] and GSE12428 [31].
For the Affymetrix data (GSE10072), Raw CEL files were processed through Affymetrix's Microarray Array Suite (MAS) 5.0 algorithm in the "affy" package in Bioconductor [32, 33]. Briefly, MAS 5.0 is a three step process which involves a global background signal correction, correction of the probe value for cross-hybridization and spurious signals using mismatch probes which are off by one base, and finally, scale normalization of each experiment to a fixed median intensity to facilitate inter-experimental comparison http://media.affymetrix.com/support/technical/whitepapers/sadd_whitepaper.pdf. Probes were filtered on MAS 5.0 calls, and those having a "P" or "M" call in at least 50% of samples were retained. This resulted in a dataset of 11440 probes. Of the 15 lung NEPS reference genes, 12 were represented on the array platform. Of those 12 genes, probes which had a "P" call in 100% of the samples were used for the calculation of the scaling factor with only one probe/gene allowed. If two probes met these criteria for one gene, the probe with the highest mean expression was chosen. After employing these criteria, eight probes were used (Additional file 5), which represented genes PPP1CB, B2M, RPL4, CAPZB, ATP5J, RAB5C, NDUFA1, and HSPA1A.
For the Agilent microarray data (GSE12428), all lung NEPS genes were represented on this microarray platform. Data was processed as described previously [31]. In the cases where lung NEPS genes were represented with multiple probes, the probe with the maximum average intensity across the dataset was used. A list of the probes used is given in Additional file 6. Since each sample had at least two replicate experiments, the average across replicate experiments was used for each probe.
To determine the scaling factor, for each sample, linear regression analysis was performed comparing the values for the reference gene (x) versus the average values for the reference genes across the sample set (y). The slope of the line based on least-squares fitting was then multiplied to each value in the experiment.
Next, Significance Analysis of Microarrays (SAM) was performed to determine differentially expressed genes between non-malignant and malignant samples for both microarray datasets using the "samr" package in R [34]. Unpaired analysis was performed using the normal samples versus tumor samples and the delta parameter set to 0.4. Probes which had a Q-value% ≤ 5 were considered significant. For the Affymetrix dataset, results were compared between the dataset normalized with MAS 5.0 alone and MAS 5.0 + NEPS scaling and for the Agilent dataset, the comparison was done between median normalization alone and NEPS scaling followed by median normalization.
Results and Discussion
Identification of reference genes for gene expression analysis in lung cancer
From our NEPS analysis [with an imposed permutation score (PS) threshold ≤ 0.15, and an average expression of ≥ 25 raw tag counts across all samples], 15 genes were identified as the most consistently expressed across normal and cancerous lung tissue (Table 1). Here we identified beta-2-microglobulin (B2M), components of the large ribosomal subunit such as ribosomal protein L19 (RPL19) and ribosomal protein L4 (RPL4), components of the small ribosomal subunit such as ribosomal protein S18 (RPS18), and electron transport chain constituents such as NADH dehydrogenase (ubiquinone) 1 alpha subcomplex 1 (NDUFA1), to rate highly in our permutation analysis, thereby suggesting their potential as reliable reference genes. B2 M has previously been utilized as a reference gene [10, 15], providing validity to the approach used here. The 18 S and 28 S rRNAs have previously served as reference genes [4, 7, 11, 12, 16], and here we show that the ribosomal protein genes can also provide this service.
Performance of standard and previously reported reference genes
A previous study reported a meta-analysis of microarray data designed to identify novel reference genes for the study of non-small cell lung cancer (NSCLC) [11]. Using a small panel of tumor/normal specimens, the authors demonstrated that genes commonly used for reference in qPCR experimentation were sub-optimal, and identified novel, more consistently expressed genes to be superior as reference genes. Additionally, studies of asthmatic airways have also shown that traditional reference genes such as ACTB and GAPDH perform poorly in this regard [6, 7, 12, 14, 15]. We find similar results using our NEPS-based approach. As described above, genes such as B2 M (permutation score = 0.011) and RPL19 (0.082) were shown to have very low permutation scores denoting stable expression between normal and cancer, whereas ACTB (2.69) and GAPDH (6.48) performed very poorly with significant differential expression. Other known reference genes such as hypoxanthine phosphoribosyltransferase 1 (HPRT1) (0.114) and TATA box binding protein (TBP) (0.468), while exhibiting low permutation scores, were not as highly expressed with average raw tag counts across all samples of 1.54 and 1.65, respectively. In contrast, genes such as peptidylprolyl isomerase A (PPIA, aka cyclophilin A) (6.11), transferrin receptor (TFRC, p90, CD71) (4.70), and phosphoglycerate kinase 1 (PGK1) (5.04) identified in a microarray meta-analysis study (see above) [11], performed poorly in our study, as revealed by the relatively high permutation scores. Although these particular genes did not perform as well as the reference genes identified from our permutation analysis, other genes identified by Saviozzi et al., such as signal transducer and activator of transcription 1 (STAT1) (0.21), esterase D/formylglutathione hydrolase (ESD) (0.18), Yes-associated protein 1 (YAP1) (0.65) and polymerase (RNA) II (DNA directed) polypeptide A (POLR2A) (0.88) did perform satisfactorily in our study, as evidenced by permutation scores ≤ 1. In addition to these genes, a second set of genes identified using a cross-tissue and cross-platform analysis were also assessed [20] and similarly, while some genes such as C-terminal binding protein 1 (CTBP1), cullin 1 (CUL1), DIM1 dimethyladenosine transferase 1-like (DIMT1L), tripartite motif-containing 27 (TRIM27) and ubiquilin 1 (UBQLN1) performed reasonably well based on our metric, others such as poly(A) polymerase alpha (PAPOLA) and ADP-ribosylation factor-like 8B (ARL8B) did not (Figure 1, Additional file 7).

Enhanced performance of the lung- NEPS genes (red; see Table 1) relative to previously reported and standard reference genes traditionally used in qPCR and microarray normalization (blue). The x-axis represents the permutation score of a defined gene and the y-axis represents the average raw (non-normalized) tag count for the same gene. Data used in the graph are given in Additional file 7. Lung NEPS genes are stable and highly expressed as compared to the traditionally used genes. B2 M appears to perform the best with respect to high average tag count and low permutation score. Notably, the gene that performs the poorest is GAPDH.
Demonstrating tissue specificity of reference genes
To further our investigations regarding reference genes optimal for cancer cell biology, we expanded our analysis to include publicly available SAGE libraries representing normal and cancer tissue from both brain and breast. The results of this analysis clearly demonstrate that the reference genes identified in the lung dataset are distinct from those found in either breast (Table 2) or brain (Table 3). This data strongly suggests that reference genes should be selected in a tissue specific manner. For example, GAPDH, which performed poorly as a reference gene for lung gene expression analysis (see above), was in fact one of the best reference genes identified from the analysis of the brain dataset (Table 3). Moreover, not only was there no overlap among the reference gene lists determined for each of the three different tissue types (i.e., lung-NEPS, breast-NEPS, brain-NEPS), but when examining reference genes specific to one tissue type (i.e. lung-NEPS) in the other two tissue types (i.e. breast or brain), the permutation scores for these genes were significantly higher and more variable (Figure 2). These results are consistent with other studies demonstrating the need for tissue and context-specific selection of reference genes [3, 5, 8, 10, 14].

Tissue-specificity of reference genes. Comparison of the permutation scores for reference genes generated in one tissue type with permutation scores for the same genes in the other two tissue types. (A) Performance of lung-NEPS genes in breast and brain tissues, (B) Performance of breast-NEPS genes in lung and brain tissues, and (C) Performance of brain-NEPS genes in lung and breast tissues.
Quantitative RT-PCR validation of identified reference genes in lung cancer samples
Using a secondary set of 15 tumor and matched non-malignant samples, qPCR was used to validate consistency of expression for all lung-NEPS genes. Additionally, we performed qPCR for previously identified, commonly used housekeeping genes ACTB, GAPDH, HPRT1, and TBP. In addition, two genes out of 13 identified (CUL1 and TRIM27) as suitable reference genes from a previously published study [20], were selected here based on high NEPS performance (see above), for qPCR analysis.
Of the NEPS genes analyzed, NDUFA1, RPL19, RAB5C, member RAS oncogene family (RAB5C), and RPS18 performed the best based on the cumulative ranking metric (Table 4). Conversely, the standard reference genes ACTB, GAPDH, and HPRT1 did not perform as well. These results confirm a high constancy of expression for a subset of the lung-NEPS genes using an alternative method in a secondary set of samples.
Effect of reference genes on differential gene expression analysis
Using a publicly available microarray dataset (GSE10072, [30]), differential expression analysis was performed using SAM [34]. Results from SAM were compared using the dataset normalized by MAS 5.0 alone, versus the same dataset normalized by MAS 5.0 with scale normalization using the lung-NEPS reference genes represented on the microarray. We observed differences in the total number of differentially regulated genes, depending on the normalization protocol used. When MAS 5.0 + NEPS normalization was used, 5502 genes were identified as up-regulated in cancer, whereas 4798 up-regulated genes were identified using MAS 5.0 alone. With respect to down-regulated genes, 2543 were identified using MAS 5.0 + NEPS, whereas 3325 were identified using MAS 5.0 alone (Figure 3A). According to the Canonical Pathway Analysis [Ingenuity Pathway Analysis (IPA)], we observe slight differences in both the number and the significance of identified pathways between the two sets of differentially normalized microarray data (Additional file 8). For example, while both datasets identify pathways such as mitochondrial dysfunction and protein ubiquitination, analysis of the dataset normalized by MAS 5.0 + NEPS identifies pathways known to be important in lung cancer, such as Neuregulin and JAK/Stat [35], at a higher significance relative to analysis of the same dataset normalized by MAS 5.0 alone (Figure 3B). Similarly, when evaluated using an additional publicly available lung cancer microarray dataset [31], we observe slight differences between the various pathways identified from analysis of differentially expressed genes derived from a NEPS-normalized dataset versus a dataset not normalized using the lung-NEPS genes (Additional file 9, Additional file 10). These results demonstrate that the choice of reference genes used for data normalization can influence the conclusions derived from gene expression studies.

SAM and pathway analysis of a dataset normalized with and without lung NEPS genes. (A) Number of probes identified as differentially over and underexpressed between cancer and normal using SAM on the dataset with and without NEPS normalization. Venn diagram illustrates the overlap in the genes identified as well as those which are different between the two analyses. (B) Canonical pathway analysis using Ingenuity Pathway Analysis. Dark blue bars represent the results from the dataset normalized with MAS 5.0 + NEPS and light blue bars represent the results from normalization using MAS 5.0 alone. The pathways which are the most significant are the most significant in both analyses. Note that key pathways such as Neuregulin signaling and JAK/Stat are identified with higher significance when normalized using the lung NEPS genes. Such differences illustrate the impact of reference gene selection and normalization on differential gene expression analysis.
Conclusions
In this study we present a methodology based upon permutation test analysis of SAGE data, to identify reference genes that more stringently satisfy the constancy requirements crucial for accurate normalization between samples utilized in gene expression experiments. Specifically, we have identified reference genes more effective for normalization than the traditional and previously reported housekeeping genes for lung, breast, and brain cancer gene expression profiling. Furthermore, we strongly emphasize that reference genes utilized for expression profiling should be selected in a tissue specific manner. Given that this methodology utilizes sequence-based data, its utility will increase as data generated from new next-generation sequencing platforms accumulate. The usage of more appropriate reference genes will have an impact on the interpretation of existing microarray data as well as expression data generated in future studies, and potentially will shed new insight into the molecular biology of cancer.
References
Quackenbush J: Microarray data normalization and transformation. Nat Genet. 2002, 32 (Suppl): 496-501. 10.1038/ng1032.
Huggett J, Dheda K, Bustin S, Zumla A: Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. 2005, 6: 279-284. 10.1038/sj.gene.6364190.
Barber RD, Harmer DW, Coleman RA, Clark BJ: GAPDH as a housekeeping gene: analysis of GAPDH mRNA expression in a panel of 72 human tissues. Physiol Genomics. 2005, 21: 389-395. 10.1152/physiolgenomics.00025.2005.
Bas A, Forsberg G, Hammarstrom S, Hammarstrom ML: Utility of the housekeeping genes 18 S rRNA, beta-actin and glyceraldehyde-3-phosphate-dehydrogenase for normalization in real-time quantitative reverse transcriptase-polymerase chain reaction analysis of gene expression in human T lymphocytes. Scand J Immunol. 2004, 59: 566-573. 10.1111/j.0300-9475.2004.01440.x.
de Kok JB, Roelofs RW, Giesendorf BA, Pennings JL, Waas ET, Feuth T, Swinkels DW, Span PN: Normalization of gene expression measurements in tumor tissues: comparison of 13 endogenous control genes. Lab Invest. 2005, 85: 154-159.
Glare EM, Divjak M, Bailey MJ, Walters EH: beta-Actin and GAPDH housekeeping gene expression in asthmatic airways is variable and not suitable for normalising mRNA levels. Thorax. 2002, 57: 765-770. 10.1136/thorax.57.9.765.
Goidin D, Mamessier A, Staquet MJ, Schmitt D, Berthier-Vergnes O: Ribosomal 18 S RNA prevails over glyceraldehyde-3-phosphate dehydrogenase and beta-actin genes as internal standard for quantitative comparison of mRNA levels in invasive and noninvasive human melanoma cell subpopulations. Anal Biochem. 2001, 295: 17-21. 10.1006/abio.2001.5171.
Khimani AH, Mhashilkar AM, Mikulskis A, O'Malley M, Liao J, Golenko EE, Mayer P, Chada S, Killian JB, Lott ST: Housekeeping genes in cancer: normalization of array data. Biotechniques. 2005, 38: 739-745. 10.2144/05385ST04.
Lee S, Jo M, Lee J, Koh SS, Kim S: Identification of novel universal housekeeping genes by statistical analysis of microarray data. J Biochem Mol Biol. 2007, 40: 226-231.
Rubie C, Kempf K, Hans J, Su T, Tilton B, Georg T, Brittner B, Ludwig B, Schilling M: Housekeeping gene variability in normal and cancerous colorectal, pancreatic, esophageal, gastric and hepatic tissues. Mol Cell Probes. 2005, 19: 101-109. 10.1016/j.mcp.2004.10.001.
Saviozzi S, Cordero F, Lo Iacono M, Novello S, Scagliotti GV, Calogero RA: Selection of suitable reference genes for accurate normalization of gene expression profile studies in non-small cell lung cancer. BMC Cancer. 2006, 6: 200-10.1186/1471-2407-6-200.
Steele BK, Meyers C, Ozbun MA: Variable expression of some "housekeeping" genes during human keratinocyte differentiation. Anal Biochem. 2002, 307: 341-347. 10.1016/S0003-2697(02)00045-3.
Szabo A, Perou CM, Karaca M, Perreard L, Quackenbush JF, Bernard PS: Statistical modeling for selecting housekeeper genes. Genome Biol. 2004, 5: R59-10.1186/gb-2004-5-8-r59.
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F: Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3: RESEARCH0034-10.1186/gb-2002-3-7-research0034.
Zhang X, Ding L, Sandford AJ: Selection of reference genes for gene expression studies in human neutrophils by real-time PCR. BMC Mol Biol. 2005, 6: 4-10.1186/1471-2199-6-4.
Zhong H, Simons JW: Direct comparison of GAPDH, beta-actin, cyclophilin, and 28 S rRNA as internal standards for quantifying RNA levels under hypoxia. Biochem Biophys Res Commun. 1999, 259: 523-526. 10.1006/bbrc.1999.0815.
Eickhoff B, Korn B, Schick M, Poustka A, van der Bosch J: Normalization of array hybridization experiments in differential gene expression analysis. Nucleic Acids Res. 1999, 27: e33-10.1093/nar/27.22.e33.
Zhu LJ, Altmann SW: mRNA and 18S-RNA coapplication-reverse transcription for quantitative gene expression analysis. Anal Biochem. 2005, 345: 102-109. 10.1016/j.ab.2005.07.028.
Jin P, Zhao Y, Ngalame Y, Panelli MC, Nagorsen D, Monsurro V, Smith K, Hu N, Su H, Taylor PR, et al: Selection and validation of endogenous reference genes using a high throughput approach. BMC Genomics. 2004, 5: 55-10.1186/1471-2164-5-55.
Kwon MJ, Oh E, Lee S, Roh MR, Kim SE, Lee Y, Choi YL, In YH, Park T, Koh SS, Shin YK: Identification of novel reference genes using multiplatform expression data and their validation for quantitative gene expression analysis. PLoS One. 2009, 4: e6162-10.1371/journal.pone.0006162.
de Jonge HJ, Fehrmann RS, de Bont ES, Hofstra RM, Gerbens F, Kamps WA, de Vries EG, van der Zee AG, te Meerman GJ, ter Elst A: Evidence based selection of housekeeping genes. PLoS One. 2007, 2: e898-10.1371/journal.pone.0000898.
Velculescu VE, Zhang L, Vogelstein B, Kinzler KW: Serial analysis of gene expression. Science. 1995, 270: 484-487. 10.1126/science.270.5235.484.
Good P: Permutation Tests: A Practical Guide to Resampling Methods for Testing Hypotheses. 2000, Springer-Verlag New York, Inc, 2
Lonergan KM, Chari R, Coe BP, Wilson IM, Tsao MS, Ng RT, Macaulay C, Lam S, Lam WL: Transcriptome profiles of carcinoma-in-situ and invasive non-small cell lung cancer as revealed by SAGE. PLoS One. 2010, 5: e9162-10.1371/journal.pone.0009162.
Lonergan KM, Chari R, Deleeuw RJ, Shadeo A, Chi B, Tsao MS, Jones S, Marra M, Ling V, Ng R, et al: Identification of novel lung genes in bronchial epithelium by serial analysis of gene expression. Am J Respir Cell Mol Biol. 2006, 35: 651-661. 10.1165/rcmb.2006-0056OC.
Chari R, Lonergan KM, Ng RT, MacAulay C, Lam WL, Lam S: Effect of active smoking on the human bronchial epithelium transcriptome. BMC Genomics. 2007, 8: 297-10.1186/1471-2164-8-297.
Boon K, Osorio EC, Greenhut SF, Schaefer CF, Shoemaker J, Polyak K, Morin PJ, Buetow KH, Strausberg RL, De Souza SJ, Riggins GJ: An anatomy of normal and malignant gene expression. Proc Natl Acad Sci USA. 2002, 99: 11287-11292. 10.1073/pnas.152324199.
Riggins GJ, Strausberg RL: Genome and genetic resources from the Cancer Genome Anatomy Project. Hum Mol Genet. 2001, 10: 663-667. 10.1093/hmg/10.7.663.
Andersen CL, Jensen JL, Orntoft TF: Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64: 5245-5250. 10.1158/0008-5472.CAN-04-0496.
Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, Dasgupta A, Mann FE, Fukuoka J, Hames M, Bergen AW, et al: Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One. 2008, 3: e1651-10.1371/journal.pone.0001651.
Boelens MC, van den Berg A, Fehrmann RS, Geerlings M, de Jong WK, te Meerman GJ, Sietsma H, Timens W, Postma DS, Groen HJ: Current smoking-specific gene expression signature in normal bronchial epithelium is enhanced in squamous cell lung cancer. J Pathol. 2009, 218: 182-191. 10.1002/path.2520.
Gautier L, Cope L, Bolstad BM, Irizarry RA: affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004, 20: 307-315. 10.1093/bioinformatics/btg405.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al: Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004, 5: R80-10.1186/gb-2004-5-10-r80.
Tusher VG, Tibshirani R, Chu G: Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001, 98: 5116-5121. 10.1073/pnas.091062498.
Liu J, Kern JA: Neuregulin-1 activates the JAK-STAT pathway and regulates lung epithelial cell proliferation. Am J Respir Cell Mol Biol. 2002, 27: 306-313.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1755-8794/3/32/prepub
Acknowledgements
We thank Drs. William W. Lockwood and Ian M. Wilson for useful discussion and editing. This work was supported by funds from Canadian Institutes of Health Research. RC is supported by scholarships from the Michael Smith Foundation for Health Research and Canadian Institutes of Health Research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
RC analyzed the SAGE and quantitative PCR data to identify the key findings, and wrote the manuscript. KML led the construction of all lung SAGE libraries, contributed to interpretation of data and manuscript editing. LAP performed the quantitative PCR experiments and data interpretation. BPC and CEM provided insight into the statistical analysis as well as manuscript editing. CQZ contributed to the experimental work. THWC contributed to the data analysis. SL isolated the clinical samples, contributed to interpretation of results. CEM, MST, SL, RTN and WLL are the principal investigators on the project. They contributed to the overall design and data interpretation. All authors have read and approved the final manuscript.
Raymond T Ng and Wan L Lam contributed equally to this work.
Electronic supplementary material
12920_2010_167_MOESM1_ESM.XLS
Additional file 1: Summary of lung SAGE libraries. Raw tag count summary for lung SAGE libraries used in analysis. (XLS 25 KB)
12920_2010_167_MOESM2_ESM.XLS
Additional file 2: Summary of breast SAGE libraries. Raw tag count summary for breast SAGE libraries used in analysis. (XLS 18 KB)
12920_2010_167_MOESM3_ESM.XLS
Additional file 3: Summary of brain SAGE libraries. Raw tag count summary for brain SAGE libraries used in analysis. (XLS 18 KB)
12920_2010_167_MOESM4_ESM.XLS
Additional file 4: Taqman probe IDs of genes assessed by quantitative RT-PCR. Applied Biosystems Taqman probe IDs for genes assessed by qRT-PCR. (XLS 26 KB)
12920_2010_167_MOESM5_ESM.XLS
Additional file 5: Probes used for NEPS genes represented on the Affymetrix U133A microarray. Probes for NEPS genes represented on the Affymetrix U133A microarray. (XLS 26 KB)
12920_2010_167_MOESM6_ESM.XLS
Additional file 6: Probes used for NEPS genes represented on the Agilent microarray. Probes used for NEPS genes represented on the Agilent microarray. (XLS 26 KB)
12920_2010_167_MOESM7_ESM.XLS
Additional file 7: Permutation scores of previously identified and NEPS identified reference genes. Data used in scatter plot shown in Figure 1. (XLS 28 KB)
12920_2010_167_MOESM8_ESM.XLS
Additional file 8: Ingenuity Pathway Analysis using genes from the analyses of a NEPS-normalized and unnormalized dataset by Landi et al. Ingenuity Pathway Analysis using genes from the analyses of a NEPS-normalized and unnormalized dataset by Landi et al. (XLS 25 KB)
12920_2010_167_MOESM9_ESM.PDF
Additional file 9: SAM and pathway analysis of an Agilent lung cancer microarray dataset normalized with and without lung NEPS genes. SAM and pathway analysis of a dataset normalized with and without lung NEPS genes. (A) Number of probes identified as differentially over and underexpressed between cancer and normal using SAM on the dataset with and without NEPS normalization. Venn diagram illustrates the overlap in the genes identified as well as those which are different between the two analyses. (B) Canonical pathway analysis using Ingenuity Pathway Analysis. Dark blue bars represent the results from the dataset normalized with NEPS and median normalization and light blue bars represent the results from using median normalization alone. While similar pathways are statistically significant, each pathway is slightly different in the degree of statistical significance. Such differences illustrate the impact of reference gene selection and normalization on differential gene expression analysis. (PDF 1 MB)
12920_2010_167_MOESM10_ESM.XLS
Additional file 10: Ingenuity Pathway Analysis using genes from the analyses of a NEPS-normalized and unnormalized dataset by Boelens et al. Ingenuity Pathway Analysis using genes from the analyses of a NEPS-normalized and unnormalized dataset by Boelens et al. (XLS 40 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chari, R., Lonergan, K.M., Pikor, L.A. et al. A sequence-based approach to identify reference genes for gene expression analysis. BMC Med Genomics 3, 32 (2010). https://doi.org/10.1186/1755-8794-3-32
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8794-3-32