- Research
- Open access
- Published:
Intellectual disability associated with craniofacial dysmorphism due to POLR3B mutation and defect in spliceosomal machinery
BMC Medical Genomics volume 15, Article number: 89 (2022)
Abstract
Background
Intellectual disability (ID) is a clinically important disease and a most prevalent neurodevelopmental disorder. The etiology and pathogenesis of ID are poorly recognized. Exome sequencing revealed a homozygous missense mutation in the POLR3B gene in a consanguineous family with three Intellectual disability with craniofacial anomalies patients. POLR3B gene encoding the second largest subunit of RNA polymerase III.
Methods
We performed RNA sequencing on blood samples to obtain insights into the biological pathways influenced by POLR3B mutation. We applied the results of our RNA-Seq analysis to several gene ontology programs such as ToppGene, Enrichr, KEGG.
Results
A significant decrease in expression of several spliceosomal RNAs, ribosomal proteins, and transcription factors was detected in the affected, compared to unaffected, family members.
Conclusions
We hypothesize that POLR3B mutation dysregulates the expression of some important transcription factors, ribosomal and spliceosomal genes, and impairments in protein synthesis and splicing mediated in part by transcription factors such as FOXC2 and GATA1 contribute to impaired neuronal function and concurrence of intellectual disability and craniofacial anomalies in our patients. Our study highlights the emerging role of the spliceosome and ribosomal proteins in intellectual disability.
Background
Intellectual disability (ID), a complex neurodevelopmental disorder, is defined as a notable impairment in cognitive and adaptive behavior before 18 years [1]. This condition, which affects approximately 1 to 3% of the general population, is a major health care problem of all developed countries. The etiology of ID can be divided into non-genetic and genetic insults. Non-genetic insults include a variety of environmental factors such as malnutrition, infection, trauma or head injury, and teratogens [2]. Most of these factors impose their effects during prenatal life [3]. Chromosomal abnormalities, dysregulation of genetic imprinting, and monogenic disease forms are significant contributors to ID [4].
Over the past 10 years, investigators have taken advantage of next-generation sequencing (NGS) technologies to identify many ID associated genes [5]. NGS is now being applied to analyze transcriptomes termed RNA-seq [6]. RNA-seq has played an important role in studying gene expression and identifying novel RNA species [7].
Although the number of genes responsible for ID increases rapidly, understanding the related processes is a challenge of basic and medical sciences. Abundant investigations have been applied to the study of the human brain based on the identification of genes implicated in ID [4]. Many of these genes in terms of modules interact together and have functional correlations that have been implicated in ID [8]. Some important molecular and biological mechanisms underlying ID have been recognized, including neurogenesis, synaptic structure and functions, immune system, and transcription and translation control [9].
A challenging area in intellectual disability is our poor understanding of the relationships among genes and how disruption of one gene affects that network and influences phenotype. Differential expression analysis is one method that can address this issue by deciphering the long lists of differentially expressed genes through combining them with other functional and ontological data [10].
RNA polymerase III (Pol III) is one of the three eukaryotic RNA polymerases. Pol III comprises 17 subunits with high conservation [11]. POLR3A and POLR3B, the two largest subunits of Pol III, encode the catalytic center of the enzyme [12]. Pol III is responsible for the synthesis of non-coding RNAs including 7SK RNA, Alu RNA, U6 RNA, H1 RNA, tRNA, 5S RNA, which are involved in cellular processes such as regulation of transcription, RNA processing, and translation [13]. Pol III plays a pivotal role in cellular processes and several studies have addressed the overall consequences of its dysfunction in mammalian cells [14, 15].
Mutations in POLR3A and POLR3B have been implicated in ID, which generally presents with 4H leukodystrophy. Recently, a study reported six unrelated individuals with de novo missense variants in POLR3B gene and clinical presentation of substantially different from POLR3-related leukodystrophy includes afferent ataxia, spasticity, variable intellectual disability and epilepsy, and predominantly demyelinating sensory-motor peripheral neuropathy [16].
This paper aims to identify differentially expressed genes and pathways in ID patients with a mutation in the POLR3B gene. We performed transcriptome analysis using RNA-sequencing on human blood cells carrying the POLR3B mutation.
Methods
This study was conducted according to the declaration of Helsinki and with the approval of the ethics board of the University of Tehran. Participants consented to participate after being informed of the nature of the research.
Subjects
The consanguineous pedigree with three ID affected members was recruited (Fig. 1, Table 1). Transcriptome analysis was performed on two affected and six unaffected individuals of this family.
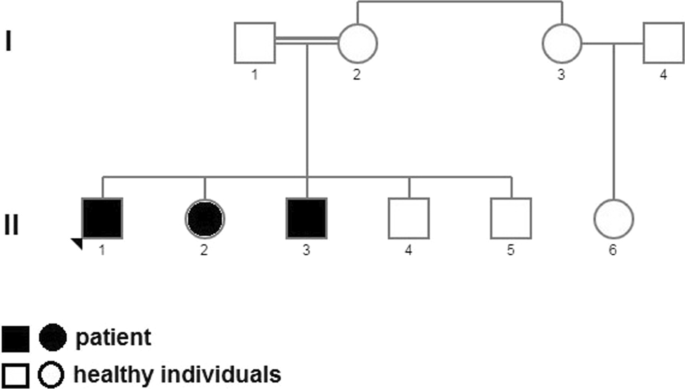
Pedigree of a family with more than two affected persons due to a homozygous missense mutation in POLR3B. II-1 is proband. II-2, II-3, II-4, II-5, II-6 and three healthy cousins (sex and age-matched) involved in this study
RNA sequencing
Blood samples were collected from eight individuals mentioned above. Total RNA was extracted from the blood by the QIAamp RNA Blood Kit (Qiagen) following the manufacturer's instructions. RNA-seq libraries were generated using Illumina TruSeq stranded total RNA with ribo-zero globin Sample Prep kit. rRNA and globin RNA were exhausted using Illumina Globin Removal Mix. The RNA was fragmented into short pieces following the purification steps using RNA Fragmentation Reagents (Life Technologies). Under these conditions, fragment lengths range from 200 to 300 bp.
The Superscript II Reverse Transcriptase and random hexamers (Life Technologies) generated the first-strand complementary DNA. The second strand was synthesized using DNA Polymerase I and RNaseH. A single ‘A” base was added to the 3’ end, followed by ligation of the Illumina sequencing PE adapters. These products were then purified and enriched by polymerase chain reaction on the adapter-ligated cDNA with 2X Phusion DNA polymerase Master Mix (New England Biolabs). 10 µg of total RNA was used to generate index-inserted paired-end cDNA libraries. Finally, RNA samples were sequenced 100 bp (2 × 100) paired-end on Illumina HiSeq2500 (Macrogen).
Data process
After obtaining the short reads, Sequence reads' quality from each sample was checked by FASTQC. Trimmomatic (v0.36) [17] was used to eliminate adaptors and low-quality bases. The ultrafast aligner Spliced Transcripts Alignment to a Reference software (STAR, v. 2.5.3) [18] was used to align all reads independently to a reference human genome assembly hg19 with the Illumina-supplied hg19 gene-model annotation file (gtf annotation). The mapped sequences were evaluated with FASTQC to ensure no artificial fragment representation. The output SAM files using SAMtools [19] were converted to BAM files, sorted by index. HTSeq-count (version 0.5.3p9) [20], a Python-based script, was used to calculate the number of aligned reads per gene.
Differential expression analysis
To identify differentially expressed genes between the patients and the healthy controls, The DESeq2 (version 1.1.0, http://www.bioconductor.org/packages/release/bioc/html/ DESeq2.html) [21] package and Cufflinks (http://cufflinks.cbcb.umd.edu) [22] were used. Expression levels of all transcripts were normalized according to the fragments per kilobase of exon per million fragments mapped (FPKM) using Cufflinks. We used the filtering criteria, including p-value of ≤ 0.05 and fold change of ≥ 1.5 to categorize transcripts as significant differentially expressed genes (DEGs).
Gene enrichment and pathway analysis
To assess the Functional annotation of the identified DEGs, pathway enrichment analysis for the DEGs (the intersection of DEGs from DESeq2 and Cufflinks) were conducted using several bioinformatics tools: ToppGene (https://toppgene.cchmc.org) [23], DAVID (https://david.ncifcrf.gov/) [24], Functional Enrichment analysis tool (FunRich v 3.1.3) [25], and Enrichr (http://amp.pharm.mssm.edu/Enrichr/) [26]. We selected pathways to adjust P-values of < 0.01 calculated by the Benjamini–Hochberg method implemented in these web tools.
Protein–protein interaction analysis and transcriptional regulators
Brain protein–protein interactome (PPI) network of the proteins encoded by the DEGs was built using NetworkAnalyst [27]. NetworkAnalyst is a comprehensive online platform for visualization and gene expression data analysis is based on experimental studies and computational predictions. It was used to find crucial modules. Genes with enormous connections in the module are often hub genes, which may have essential functions.
Also, to find interactions, the JASPER database [28] for DEGs-TFs and miRTarBase v8.0 [29] database for Gene-miRNAs in NetworkAnalyst were applied to generating related networks.
Comparison of our significant DEGs with genes identified in a mouse with mutation in Polr3b R103H causing Leukodystrophy
POLR3A and POLR3B protein sequences have great conservation between humans and mice. Bernard Brais et al. using mice models, have studied POLR3-related hypomyelinating leukodystrophy (POLR3-HLD). They found that Polr3a G672E homozygote mutation had no neurological deficits, and Polr3b R103H homozygote mutation was embryonically lethal.
Polr3aG672E/G672E/Polr3b+/R103H double mutant mice were generated. Then, three affected mice were compared to three healthy mice using RNA-seq [30]. Here, using data from the Gene expression omnibus (GSE118739), we compared the DEGs in Polr3b mutated mouse and DEGs in our POLR3B mutated patients. DEGs were identified using DESeq2 (version 1.1.0) in the same way done for our gene list. Biomart was used to convert mouse gene IDs to their orthologous IDs in humans, then, DEGs in mice and DEGs in our ID patients were compared.
Results
POLR3B mutated family
There were eight participants in this study. Four of them are from a consanguineous family, including two patients and two controls. Figure 1 shows the family pedigree. Four other samples are healthy cousins matched by age and sex, which have added to more precise patient-control collation results.
Previously, Whole Exome Sequencing identified a homozygous missense mutation NM_018082.4:c.770C > A; p.(Thr257Lys)) in POLR3B gene [31]. The variant allele was completely absent in healthy controls in this study. POLR3B encodes one of the core components of RNA polymerase III, which transcripts small RNAs U2 and 5S rRNAs. This mutation caused severe intellectual disability, attention deficit, and autistic behavior with facial dysmorphism in three patients from first cousin healthy parents. Their facial appearances showed long palpebral fissures, flat occiput, short philtrum, protrude ear, and micrognathia.
Brain MRI of the oldest patient had hypomyelinating leukodystrophy. Cognitive status which was evaluated using WAIS-IV in three patients showed IQs of 25–40, in the range of severe ID. Table 1 details the phenotypes of the affected individuals.
Differential expression analysis
Cuffdiff from the cufflinks package identified differential expressed genes between two patients and six healthy controls. We considered genes as DEGs with the parameter of p-value less than 0.05. We detected 532 differentially expressed genes, with 311 genes downregulated and 221 genes upregulated in ID patients compared to controls (Additional file 1: Table S1, Fig. 2a).

Differentially expressed genes between POLR3B mutant patients and 6 controls. a The heat map showing DEGs between POLR3B mutant patients vs controls. b Comparison of POLR3B expression in POLR3B mutant patients and controls. c Comparison of POLR3B isoform expression in POLR3B mutant patients and controls
Among the downregulated genes, the expression value of 27 genes was zero in the patients; most of the genes are coding small nuclear RNAs (Table 2). Enrichment analysis for these 27 genes using the ToppGene site indicated pre-mRNA 5'-splice site binding (GO:0,030,627) as the primary Molecular Function (Table 3). From these 27 genes, 10 genes participate in spliceosome structure and mRNA splicing. For example, binding of U1 snRNA to the 5’ splice site is necessary for spliceosome assembly [32]. RNU11 gene belongs to the snRNA class, and the mutation in this gene is associated with Microcephalic Osteodysplastic Primordial Dwarfism, Type I [33].
Among the upregulated genes, the expression value of 7 genes was zero in the controls (Table 2). Between them, NME1-NME2 was the only protein-coding gene (Table 2). NME1-NME2 are parts of the NME gene family with ten members. This locus represents naturally occurring read-through transcription between the neighboring NME1 and NME2 genes. Depending on tissue context, both have a crucial role in tumor progression and metastasis [34]. Recently, a study published in Psychiatric Genetics has represented a homozygous mutation in this gene can be associated with attention deficit hyperactivity disorder (ADHD) [35].
Our results revealed that the mutation didn’t change the expression level of POLR3B, so it presumably alters the function of its protein (Fig. 2b). POLR3B has four isoforms, and measurements show no significant difference between patients and controls (Fig. 2c).
The top 10 down-regulated genes in the patients are listed in Table 4 and Fig. 3. Of these, the SLC12A1 gene had the greatest fold change. SLC12A1 encodes solute carrier family 12 members 1 protein and is implicated in ID in the literature [36]. The top 10 up-regulated genes in the patients are listed in Table 4 and Fig. 3. The GAD1 gene with the greatest fold change was reported as a causative gene associated with syndromic developmental and epileptic encephalopathy [37]. Among DEGs, 30 genes were reported as an ID gene [38] (Table 5). Of these, 21 genes were down-regulated in the patients.

The top 10 down-regulated and up-regulated genes in POLR3B mutant patients vs controls
Pathway analysis
To assess the biological process and significant molecular mechanisms underlying the pathogenesis of ID, we analyzed the DEGs by ToppFun application of ToppGene suite in terms of molecular function, cellular component, biological process, and biological pathway. The analyses showed spliceosomal snRNP assembly and innate immune response were involved as the main biological processes (Table 6). The molecular functions and cellular components encoded by the DEGs were significantly related to the ribosome and its subunits, spliceosomal snRNP complex, and Nonsense Mediated Decay (NMD) (Table 6).
The most striking pathways related to intellectual disability were "immune system", "translation", "spliceosomal snRNP assembly", "Nonsense Mediated Decay" (Table 6).
Protein–protein interaction analysis
We used tissue-specific (cortex) protein–protein interactome data to construct protein–protein interaction (PPI) network. Several subnetworks around the DEGs were achieved, the first subnetwork had 2495 nodes, and 4169 edges contained 288 seed nodes. Then, a minimum network was applied to reconstruct a subnetwork with 749 nodes, 2044 edges, and 288 seeds. Network analyst software was applied to visualize the interaction network (Fig. 4). The degree-based topological analysis with force atlas layout showed 34 Hub genes. Additional file 1: Table S2 lists the details of the Hub proteins.

protein–protein interactions (PPI) of the DEGs. Nodes and edges represented by colored circles and arrows respectively. The big circle nodes are the hub proteins
Transcriptional regulators
We constructed a TF-Genes network-based interaction using the JASPER database. We applied the minimum network option on subnetworks to attain a unique network and filtered the result by brain tissue. The reconstructed subnetwork had 485 nodes, 3218 edges, and 411 seed nodes (Fig. 5). Transcription factors with binding sites for greater number of DEGs were FOXC1, GATA2, FOXL1, YY1, NF1C, STAT1, and ELK4 (Table 7).

Transcriptional regulators of DEGs and miRNA. Transcriptional regulators of DEGs. Diamonds represent TFs and red circles show nodes, they are related by arrows
Also, we investigated the relation of Gene-miRNAs using miRTarBase v8.0 database, which experimentally validated miRNAs-Gene interaction data. We attained a network with more than 2000 nodes, so we used a minimum network option and considered nodes with at least 10 degrees. After filtering, a subnetwork with 102 nodes, 465 edges and 33 seeds were built. PRRG4 and mir-92a-3p were the crucial node and miRNA, respectively.
Comparison of our significant degs to genes identified in a mouse with an R103H mutation in Polr3b causing Leukodystrophy
From 255 DEGs in mice, 147 genes have a homolog in humans. Among these 147 genes, 5 were shared between our POLR3B mutated patients and Polr3b mutated mice (MYL4, RAB44, LY6G6E, TRAF5, CKM). Tumor necrosis factor receptor-associated factor 5 (TRAF5) interacts with downstream effectors, including tumor necrosis factor (TNF) and interleukin-1 receptor/Toll-like receptor. TRAF5 plays key role in regulating several signaling pathways such as Nod-like receptor pathway and Akt/FoxO1 signaling pathway. It has been found neuronal apoptosis level, blood–brain barrier (BBB) degradation, and inflammatory response reduced in TRAF5 Knockout Mice. Also, TRAF5 protein expression significantly increased in ischemic brains [39].
Discussion
The advent of next-generation sequencing technologies has detected a large number of causative genes in ID. Studies of transcriptome changes in ID patients compared to healthy controls, are limited due to the difficulties in accessing tissues, here we performed a comprehensive transcriptome analysis of total RNA extracted from the blood from members of a family affected by a recessive mutation POLR3B. Our data's most significant differentially expressed pathways between patients and healthy controls were Ribosome, Nonsense-mediated decay, spliceosomal snRNP assembly, immune system.
Our results showed that numerous spliceosomal genes were significantly dysregulated in POLR3B mutant patients. The spliceosome is a large protein-RNA complex that removes introns from nuclear pre-mRNA. Researchers have revealed mutations in pre-mRNA splicing factor genes causes craniofacial anomalies [40]. Along the same lines, studies have shown mutation in components of spliceosome causes concurrence of ID, short stature, poor speech, and craniofacial anomalies [41]. Recently, Lee and colleagues have shown X-linked ID causative mutations in the FAM50A gene dysregulate the expression of spliceosomal RNAs and transcripts involved in neurodevelopment [42]. Furthermore, mutations in subunits of RNA polymerase III (POLR1D and POLR1C) have been identified in Treacher Collins syndrome (TCS), which is a malformation craniofacial disorder [43, 44]. Here, we detected a significant decrease expression of some spliceosomal RNAs (Table 2) in our ID patients. Clinical features of our patients (Table 1) shows that they have severe craniofacial anomalies. Finally, we predict that POLR3B mutation in our patients dysregulated expression of splicing factor genes and caused Intellectual disability with craniofacial anomalies in our patients.
Downregulation of numerous ribosomal proteins was also observed in POLR3B mutant patients, and ribosome was one of the most significant pathways dysregulated in the current study. Twenty-two ribosomal proteins include ribosomal S, L, and M subunits downregulated in our patients. RNA polymerase III synthesizes transfer and small ribosomal RNAs in eukaryotes. Ribosome biogenesis plays key role in regulating protein synthesis capacity in different tissues[45]. Previous studies have shown deficiency of ribosomal proteins may cause a reduction in rRNA synthesis and vice versa [46, 47]. Therefore, it seems that the downregulation of ribosomal proteins in our patients is due to POLR3B deficiency and a reduction of rRNA synthesis. In the literature, RPL10 mutations have been reported to cause neurodevelopmental disorders with the clinical spectrum from autism to syndromic ID [48, 49]. Studies have shown that several ribosomal genes were downregulated in the hippocampus of Alzheimer patients (AD)[50]. Recently, scientists recommended that ribosomal dysfunction in peripheral blood might be related to prodrome and progression of AD [51]. Therefore, downregulation of ribosomal proteins in our patients may disrupt protein synthesis and contribute to cognitive impairment.
Our DEGs include several cell adhesion molecules (CAMs) and immune system genes such as ICAM3, SELP, CLDN5, CD274, CD8B, CD8A, SDC2. CAMs play important roles in the nervous system. They control the interaction of neurons and glia, synapse formation and neurite outgrowth [52]. Three genome –wide association studies (GWAS) demonstrated aberrant CAM molecules are associated with neurological disorders, including schizophrenia and bipolar disorder[53]. Several ID and neurodevelopmental disease causative mutations in different CAMs such as L1CAM and ICAM3 have been demonstrated in different studies [54].
This study also identified the potential TFs using the topological analysis of protein–protein interactions (Table 7). They include FOXC1, GATA2, YY1, FOXL1, NFIC, PPARG, and E2F1. FOXC1 deletion or duplication can lead to cerebellar and posterior fossa malformations [55]. Also, two case report studies have also shown that ring chromosome 6 encompassing FOXC1 is associated with intellectual disability, short stature, and multiple facial dysmorphisms [56, 57]. GATA2 expression in posterior diencephalon-midbrain is crucial to GABAergic neuron development, migration, and regulation of neuron-specific gene expression [58]. Whole-Exome Sequencing revealed a mutation in GATA2 causes a rare Syndromic Congenital Neutropenia With Intellectual Disability [59]. FOXl1 is another member of Forkhead box (FOX) proteins whose dysregulation activates the Wnt/b-catenin pathway. [60] YY1 controls brain development, proliferation, and survival of neural progenitor cells (NPCs) By regulating many metabolic pathways and protein translation [61]. YY1 deletions and point mutations lead to syndromic ID with a wide variety of phenotypic features, including cognitive impairment, behavioral alterations, intrauterine growth restriction because of transcriptional and chromatin dysfunction [62]. PRRG4 is located in the 11p13 region, relevant to WAGR syndrome. Toshiyuki Yamamoto et al. suggested haploinsufficiency of PAX6 or PRRG4 caused severe developmental delay and autistic behaviors of WAGR syndrome [63].
We investigated miRNAs-Gene interaction using miRTarBase v8.0 database. As a result, PRRG4 and mir-92a-3p were the crucial node and miRNA. mir-92a-3p has an association with synaptic structure and function. Also, it was identified as a biomarker in peripheral blood for schizophrenia [64]. In addition, a study on gene regulatory associated with autism in the Chinese population showed mir-92a-3p dysregulation in the peripheral blood of patients [65].
In the current study, we have used blood transcriptome to identify differentially expressed genes and pathways in POLR3B mutant patients. Since brain tissues were not available, we performed RNA sequencing on patients’ blood samples. Several studies have shown great similarity between blood and brain transcriptome. Therefore, blood is considered good an alternate. Our results have shown that mutation in POLR3B gene dysregulated the expression of some important transcription factors and spliceosome genes. DEGs are involved in some important biological processes such as spliceosome assembly, ribosome, and NMD.
The limitations of this study
A single family with a single variant was analysed. The analysis is in blood derived RNA rather than brain. A small sample size of affected individuals were analysed thus a high number of false positive findings are expected. Inability to independently replicate the key findings using an alternative analysis of RNA expression such as quantitative real-time PCR.
Conclusion
We hypothesize that POLR3B mutation dysregulates the expression of some critical transcription factors, ribosomal and spliceosomal genes. Impairments in protein synthesis and splicing mediated in part by transcription factors such as FOXC2, GATA1, contribute to impaired neuronal function and concurrence of intellectual disability and craniofacial anomalies in our patients. Our study highlights the emerging role of the spliceosome and ribosomal proteins in intellectual disability.
Availability of data and materials
The datasets used and analysed during the current study are available in GEO with accession number GSE184234.
Abbreviations
- ID:
-
Intellectual disability
- RNA:
-
Polymerase III (Pol III)
- FPKM:
-
Fragments per kilobase of exon per million fragments mapped
- PPI:
-
Protein–protein interactome
- POLR3-HLD:
-
POLR3-related hypomyelinating leukodystrophy
- NMD:
-
Nonsense Mediated Decay
- TRAF5:
-
Tumor necrosis factor receptor-associated factor 5
- TNF:
-
Tumor necrosis factor
- BBB:
-
Blood–brain barrier
- TCS:
-
Treacher Collins syndrome
- AD:
-
Alzheimer patients
- CAMs:
-
Cell adhesion molecules
- FOX:
-
Forkhead box
- NPCs:
-
Neural progenitor cells
References
Vissers LE, Gilissen C, Veltman JA. Genetic studies in intellectual disability and related disorders. Nat Rev Genet. 2016;17(1):9.
Mir YR, Kuchay RAH. Advances in identification of genes involved in autosomal recessive intellectual disability: a brief review. J Med Genet. 2019;56(9):567–73.
Chiurazzi P, Pirozzi F. Advances in understanding–genetic basis of intellectual disability. F1000Res. 2016;5.
Gécz J. The molecular basis of intellectual disability: novel genes with naturally occurring mutations causing altered gene expression in the brain. Front Biosci. 2004;9:1–7.
Van Dijk EL, Auger H, Jaszczyszyn Y, Thermes C. Ten years of next-generation sequencing technology. Trends Genet. 2014;30(9):418–26.
Marguerat S, Bähler J. RNA-seq: from technology to biology. Cell Mol Life Sci. 2010;67(4):569–79.
Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57.
Van Bokhoven H. Genetic and epigenetic networks in intellectual disabilities. Annu Rev Genet. 2011;45:81–104.
Chelly J, Khelfaoui M, Francis F, Chérif B, Bienvenu T. Genetics and pathophysiology of mental retardation. Eur J Hum Genet. 2006;14(6):701–13.
Parikshak NN, Gandal MJ, Geschwind DH. Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat Rev Genet. 2015;16(8):441–58.
Terhal PA, Vlaar JM, Middelkamp S, Nievelstein RA, Nikkels PG, Ross J, et al. Biallelic variants in POLR3GL cause endosteal hyperostosis and oligodontia. Eur J Hum Genet. 2020;28(1):31–9.
Choquet K, Yang S, Moir RD, Forget D, Larivière R, Bouchard A, et al. Absence of neurological abnormalities in mice homozygous for the Polr3a G672E hypomyelinating leukodystrophy mutation. Mol Brain. 2017;10(1):1–13.
Dorboz I, Dumay-Odelot H, Boussaid K, Bouyacoub Y, Barreau P, Samaan S, et al. Mutation in POLR3K causes hypomyelinating leukodystrophy and abnormal ribosomal RNA regulation. Neurology Genetics. 2018;4(6).
Choquet K, Forget D, Meloche E, Dicaire M-J, Bernard G, Vanderver A, et al. Leukodystrophy-associated POLR3A mutations down-regulate the RNA polymerase III transcript and important regulatory RNA BC200. J Biol Chem. 2019;294(18):7445–59.
Lata E, Choquet K, Sagliocco F, Brais B, Bernard G, Teichmann M. RNA polymerase III subunit mutations in genetic diseases. Front Mol Biosci. 2021;8:696438.
Djordjevic D, Pinard M, Gauthier M-S, Smith-Hicks C, Hoffman TL, Wolf NI, et al. De novo variants in POLR3B cause ataxia, spasticity, and demyelinating neuropathy. Am J Hum Genet. 2021;108(1):186–93.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–9.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–78.
Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucl Acids Res. 2009;37:W305-11.
da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucl Acids Res. 2009;37(1):1–13.
Pathan M, Keerthikumar S, Ang CS, Gangoda L, Quek CY, Williamson NA, et al. FunRich: an open access standalone functional enrichment and interaction network analysis tool. Proteomics. 2015;15(15):2597–601.
Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128.
Xia J, Gill EE, Hancock REW. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat Protoc. 2015;10(6):823–44.
Wasserman WW, Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet. 2004;5(4):276–87.
Huang HY, Lin YC, Li J, Huang KY, Shrestha S, Hong HC, et al. miRTarBase 2020: updates to the experimentally validated microRNA-target interaction database. Nucleic Acids Res. 2020;48(D1):D148–54.
Choquet K, Pinard M, Yang S, Moir RD, Poitras C, Dicaire M-J, et al. The leukodystrophy mutation Polr3b R103H causes homozygote mouse embryonic lethality and impairs RNA polymerase III biogenesis. Mol Brain. 2019;12(1):59.
Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W, et al. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature. 2011;478(7367):57–63.
Eperon IC, Makarova OV, Mayeda A, Munroe SH, Cáceres JF, Hayward DG, et al. Selection of alternative 5’ splice sites: role of U1 snRNP and models for the antagonistic effects of SF2/ASF and hnRNP A1. Mol Cell Biol. 2000;20(22):8303–18.
Baumgartner M, Olthof AM, Aquino GS, Hyatt KC, Lemoine C, Drake K, et al. Minor spliceosome inactivation causes microcephaly, owing to cell cycle defects and death of self-amplifying radial glial cells. Development. 2018;145(17):dev166322.
Mezzofanti E, Ignesti M, Hsu T, Gargiulo G, Cavaliere V. Vps28 is involved in the intracellular trafficking of Awd, the Drosophila homologue of NME1/2. Front Physiol. 2019;10:983.
Shinwari JM, Al Yemni EA, Alnaemi FM, Abebe D, Al-Abdulaziz BS, Al Mubarak BR, et al. Analysis of shared homozygosity regions in Saudi siblings with attention deficit hyperactivity disorder. Psychiatr Genet. 2017;27(4):131.
Crespi B, Summers K, Dorus S. Evolutionary genomics of human intellectual disability. Evol Appl. 2010;3(1):52–63.
Chatron N, Becker F, Morsy H, Schmidts M, Hardies K, Tuysuz B, et al. Bi-allelic GAD1 variants cause a neonatal onset syndromic developmental and epileptic encephalopathy. Brain. 2020;143(5):1447–61.
Ilyas M, Mir A, Efthymiou S, Houlden H. The genetics of intellectual disability: advancing technology and gene editing. F1000Res. 2020;9:F1000 Faculty Rev-22.
Wang L, Lu Y, Guan H, Jiang D, Guan Y, Zhang X, et al. Tumor necrosis factor receptor-associated factor 5 is an essential mediator of ischemic brain infarction. J Neurochem. 2013;126(3):400–14.
Lehalle D, Wieczorek D, Zechi-Ceide RM, Passos-Bueno MR, Lyonnet S, Amiel J, et al. A review of craniofacial disorders caused by spliceosomal defects. Clin Genet. 2015;88(5):405–15.
Rangel-Sosa MM, Figuera-Villanueva LE, González-Ramos IA, Pérez-Páramo YX, Martínez-Jacobo LA, Arnaud-López L, et al. Exome sequencing reveals three homozygous missense variants in SNRPA in two sisters with syndromic intellectual disability. Clin Genet. 2018;93(6):1229–33.
Lee Y-R, Khan K, Armfield-Uhas K, Srikanth S, Thompson NA, Pardo M, et al. Mutations in FAM50A suggest that Armfield XLID syndrome is a spliceosomopathy. Nat Commun. 2020;11(1):3698.
Dauwerse JG, Dixon J, Seland S, Ruivenkamp CAL, van Haeringen A, Hoefsloot LH, et al. Mutations in genes encoding subunits of RNA polymerases I and III cause Treacher Collins syndrome. Nat Genet. 2011;43(1):20–2.
Schaefer E, Collet C, Genevieve D, Vincent M, Lohmann DR, Sanchez E, et al. Autosomal recessive POLR1D mutation with decrease of TCOF1 mRNA is responsible for Treacher Collins syndrome. Genet Med. 2014;16(9):720–4.
Thomson E, Ferreira-Cerca S, Hurt E. Eukaryotic ribosome biogenesis at a glance. J Cell Sci. 2013;126(21):4815–21.
Juli G, Gismondi A, Monteleone V, Caldarola S, Iadevaia V, Aspesi A, et al. Depletion of ribosomal protein S19 causes a reduction of rRNA synthesis. Sci Rep. 2016;6(1):35026.
Idol RA, Robledo S, Du H-Y, Crimmins DL, Wilson DB, Ladenson JH, et al. Cells depleted for RPS19, a protein associated with Diamond Blackfan Anemia, show defects in 18S ribosomal RNA synthesis and small ribosomal subunit production. Blood Cells Mol Dis. 2007;39(1):35–43.
Zanni G, Kalscheuer VM, Friedrich A, Barresi S, Alfieri P, Di Capua M, et al. A novel mutation in RPL10 (Ribosomal Protein L10) causes X-linked intellectual disability, cerebellar hypoplasia, and spondylo-epiphyseal dysplasia. Hum Mutat. 2015;36(12):1155–8.
Thevenon J, Michot C, Bole C, Nitschke P, Nizon M, Faivre L, et al. RPL10 mutation segregating in a family with X-linked syndromic Intellectual Disability. Am J Med Genet A. 2015;167(8):1908–12.
Hernández-Ortega K, Garcia-Esparcia P, Gil L, Lucas JJ, Ferrer I. Altered machinery of protein synthesis in Alzheimer’s: from the nucleolus to the ribosome. Brain Pathol. 2016;26(5):593–605.
Xue W, Li J, Fu K, Teng W. Differential expression of mRNAs in peripheral blood related to prodrome and progression of Alzheimer’s disease. Biomed Res Int. 2020;2020:4505720.
Fields RD. Cell adhesion molecules: Implications for neurological disease. Neuroscientist. 1998;4(1):4–8.
Corvin AP. Neuronal cell adhesion genes: Key players in risk for schizophrenia, bipolar disorder and other neurodevelopmental brain disorders? Cell Adh Migr. 2010;4(4):511–4.
Wong EV, Kenwrick S, Willems P, Lemmon V. Mutations in the cell adhesion molecule LI cause mental retardation. Trends Neurosci. 1995;18(4):168–72.
Aldinger KA, Lehmann OJ, Hudgins L, Chizhikov VV, Bassuk AG, Ades LC, et al. FOXC1 is required for normal cerebellar development and is a major contributor to chromosome 6p25 3 Dandy-Walker malformation. Nat Genet. 2009;41(9):1037–42.
Sheth F, Liehr T, Shah V, Shah H, Tewari S, Solanki D, et al. A child with intellectual disability and dysmorphism due to complex ring chromosome 6: identification of molecular mechanism with review of literature. Ital J Pediatr. 2018;44(1):1–9.
Pace NP, Maggouta F, Twigden M, Borg I. Molecular cytogenetic characterisation of a novel de novo ring chromosome 6 involving a terminal 6p deletion and terminal 6q duplication in the different arms of the same chromosome. Mol Cytogenet. 2017;10(1):1–6.
Shrestha S, Offer SM. Epigenetic regulations of GABAergic neurotransmission: relevance for neurological disorders and epigenetic therapy. Med Epigenet. 2016;4(1):1–19.
Gauthier-Vasserot A, Thauvin-Robinet C, Bruel AL, Duffourd Y, St-Onge J, Jouan T, et al. Application of whole-exome sequencing to unravel the molecular basis of undiagnosed syndromic congenital neutropenia with intellectual disability. Am J Med Genet A. 2017;173(1):62–71.
Benayoun BA, Caburet S, Veitia RA. Forkhead transcription factors: key players in health and disease. Trends Genet. 2011;27(6):224–32.
Zurkirchen L, Varum S, Giger S, Klug A, Häusel J, Bossart R, et al. Yin Yang 1 sustains biosynthetic demands during brain development in a stage-specific manner. Nat Commun. 2019;10(1):1–16.
Gabriele M, Vulto-van Silfhout AT, Germain P-L, Vitriolo A, Kumar R, Douglas E, et al. YY1 haploinsufficiency causes an intellectual disability syndrome featuring transcriptional and chromatin dysfunction. Am J Hum Genet. 2017;100(6):907–25.
Yamamoto T, Togawa M, Shimada S, Sangu N, Shimojima K, Okamoto N. Narrowing of the responsible region for severe developmental delay and autistic behaviors in WAGR syndrome down to 1.6 Mb including PAX6, WT1, and PRRG4. Am J Med Genet Part A. 2014;164(3):634–8.
Ma J, Shang S, Wang J, Zhang T, Nie F, Song X, et al. Identification of miR-22-3p, miR-92a-3p, and miR-137 in peripheral blood as biomarker for schizophrenia. Psychiatry Res. 2018;265:70–6.
Huang F, Long Z, Chen Z, Li J, Hu Z, Qiu R, et al. Investigation of gene regulatory networks associated with autism spectrum disorder based on MiRNA expression in China. PloS one. 2015;10(6):e0129052.
Acknowledgements
We thank patients and their family members for participating in the current study.
Funding
We acknowledge National institute for medical research development (ID: 957065) and the Genetic Research Center of University of Social Welfare and Rehabilitation Sciences for funding this research.
Author information
Authors and Affiliations
Contributions
MS performed all the experiments and wrote the draft of manuscript; KIR conceived the study and were in charge of overall direction and planning, supervised the project, wrote the manuscript. AA, KK, HN were involved in planning and supervised the work. All authors discussed the results and contributed to the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was conducted in accordance with the declaration of Helsinki and with approval of the ethics board of the University of Tehran. Participants consented to participate after being informed of the nature of the research.
Consent for publication
Not applicable.
Competing interests
The authors have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary Table 1. Dysregulated genes in POLR3B mutants patients.
Additional file 2.
Supplementary Table 2. Details of the hub genes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visithttp://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Saghi, M., InanlooRahatloo, K., Alavi, A. et al. Intellectual disability associated with craniofacial dysmorphism due to POLR3B mutation and defect in spliceosomal machinery. BMC Med Genomics 15, 89 (2022). https://doi.org/10.1186/s12920-022-01237-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-022-01237-5